Clumping GWAS Results
Introduction
After performing a genome-wide association study (GWAS) for a given phenotype, an analyst might want to clump the association results based on the correlation between variants and p-values. The goal is to get a list of independent associated loci accounting for linkage disequilibrium between variants.
For example, given a region of the genome with three variants: SNP1, SNP2, and SNP3. SNP1 has a p-value of 1e-8, SNP2 has a p-value of 1e-7, and SNP3 has a p-value of 1e-6. The correlation between SNP1 and SNP2 is 0.95, SNP1 and SNP3 is 0.8, and SNP2 and SNP3 is 0.7. We would want to report SNP1 is the most associated variant with the phenotype and “clump” SNP2 and SNP3 with the association for SNP1.
Hail is a highly flexible tool for performing analyses on genetic datasets in a parallel manner that takes advantage of a scalable compute cluster. However, LD-based clumping is one example of many algorithms that are not available in Hail, but are implemented by other bioinformatics tools such as PLINK. We use Batch to enable functionality unavailable directly in Hail while still being able to take advantage of a scalable compute cluster.
To demonstrate how to perform LD-based clumping with Batch, we’ll use the 1000 Genomes dataset from the Hail GWAS tutorial. First, we’ll write a Python Hail script that performs a GWAS for caffeine consumption and exports the results as a binary PLINK file and a TSV with the association results. Second, we’ll build a docker image containing the custom GWAS script and Hail pre-installed and then push that image to the Google Container Repository. Lastly, we’ll write a Python script that creates a Batch workflow for LD-based clumping with parallelism across chromosomes and execute it with the Batch Service. The job computation graph will look like the one depicted in the image below:
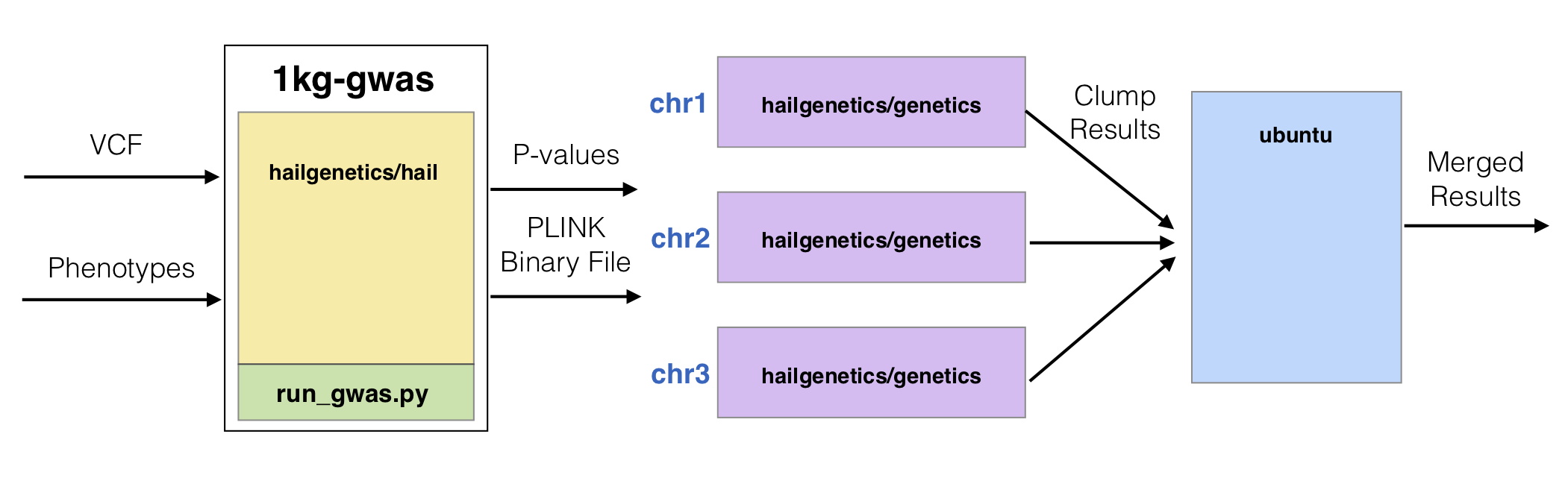
Hail GWAS Script
We wrote a stand-alone Python script run_gwas.py that takes a VCF file, a phenotypes file,
the output destination file root, and the number of cores to use as input arguments.
The Hail code for performing the GWAS is described
here.
We export two sets of files to the file root defined by --output-file. The first is
a binary PLINK file set with three files
ending in .bed, .bim, and .fam. We also export a file with two columns SNP and P which
contain the GWAS p-values per variant.
Notice the lines highlighted below. Hail will attempt to use all cores on the computer if no
defaults are given. However, with Batch, we only get a subset of the computer, so we must
explicitly specify how much resources Hail can use based on the input argument --cores.
import argparse
import hail as hl
def run_gwas(vcf_file, phenotypes_file, output_file):
table = hl.import_table(phenotypes_file, impute=True).key_by('Sample')
hl.import_vcf(vcf_file).write('tmp.mt')
mt = hl.read_matrix_table('tmp.mt')
mt = mt.annotate_cols(pheno=table[mt.s])
mt = hl.sample_qc(mt)
mt = mt.filter_cols((mt.sample_qc.dp_stats.mean >= 4) & (mt.sample_qc.call_rate >= 0.97))
ab = mt.AD[1] / hl.sum(mt.AD)
filter_condition_ab = (
(mt.GT.is_hom_ref() & (ab <= 0.1))
| (mt.GT.is_het() & (ab >= 0.25) & (ab <= 0.75))
| (mt.GT.is_hom_var() & (ab >= 0.9))
)
mt = mt.filter_entries(filter_condition_ab)
mt = hl.variant_qc(mt)
mt = mt.filter_rows(mt.variant_qc.AF[1] > 0.01)
eigenvalues, pcs, _ = hl.hwe_normalized_pca(mt.GT)
mt = mt.annotate_cols(scores=pcs[mt.s].scores)
gwas = hl.linear_regression_rows(
y=mt.pheno.CaffeineConsumption,
x=mt.GT.n_alt_alleles(),
covariates=[1.0, mt.pheno.isFemale, mt.scores[0], mt.scores[1], mt.scores[2]],
)
gwas = gwas.select(SNP=hl.variant_str(gwas.locus, gwas.alleles), P=gwas.p_value)
gwas = gwas.key_by(gwas.SNP)
gwas = gwas.select(gwas.P)
gwas.export(f'{output_file}.assoc', header=True)
hl.export_plink(mt, output_file, fam_id=mt.s, ind_id=mt.s)
if __name__ == '__main__':
parser = argparse.ArgumentParser()
parser.add_argument('--vcf', required=True)
parser.add_argument('--phenotypes', required=True)
parser.add_argument('--output-file', required=True)
parser.add_argument('--cores', required=False)
args = parser.parse_args()
if args.cores:
hl.init(master=f'local[{args.cores}]')
run_gwas(args.vcf, args.phenotypes, args.output_file)
Docker Image
A Python script alone does not define its dependencies such as on third-party packages. For
example, to execute the run_gwas.py script above, Hail must be installed as well as the
libraries Hail depends on. Batch uses Docker images to define these dependencies including
the type of operating system and any third-party software dependencies. The Hail team maintains a
Docker image, hailgenetics/hail, for public use with Hail already installed. We extend this
Docker image to include the run_gwas.py script.
FROM hailgenetics/hail:0.2.37
COPY run_gwas.py /
The following Docker command builds this image:
docker pull hailgenetics/hail:0.2.37
docker build -t 1kg-gwas -f Dockerfile .
Batch can only access images pushed to a Docker repository. You have two repositories available to you: the public Docker Hub repository and your project’s private Google Container Repository (GCR). It is not advisable to put credentials inside any Docker image, even if it is only pushed to a private repository.
The following Docker command pushes the image to GCR:
docker tag 1kg-gwas us-docker.pkg.dev/<MY_PROJECT>/1kg-gwas
docker push us-docker.pkg.dev/<MY_PROJECT>/1kg-gwas
Replace <MY_PROJECT> with the name of your Google project. Ensure your Batch service account
can access images in GCR.
Batch Script
The next thing we want to do is write a Hail Batch script to execute LD-based clumping of association results for the 1000 genomes dataset.
Functions
GWAS
To start, we will write a function that creates a new Job on an existing Batch that
takes as arguments the VCF file and the phenotypes file. The return value of this
function is the Job that is created in the function, which can be used later to
access the binary PLINK file output and association results in downstream jobs.
def gwas(batch, vcf, phenotypes):
"""
QC data and get association test statistics
"""
cores = 2
g = batch.new_job(name='run-gwas')
g.image('us-docker.pkg.dev/<MY_PROJECT>/1kg-gwas:latest')
g.cpu(cores)
g.declare_resource_group(ofile={
'bed': '{root}.bed',
'bim': '{root}.bim',
'fam': '{root}.fam',
'assoc': '{root}.assoc'
})
g.command(f'''
python3 /run_gwas.py \
--vcf {vcf} \
--phenotypes {phenotypes} \
--output-file {g.ofile} \
--cores {cores}
''')
return g
A couple of things to note about this function:
The image is the image created in the previous step. We copied the run_gwas.py script into the root directory /. Therefore, to execute the run_gwas.py script, we call /run_gwas.py.
The run_gwas.py script takes an output-file parameter and then creates files ending with the extensions .bed, .bim, .fam, and .assoc. In order for Batch to know the script is creating files as a group with a common file root, we need to use the
BashJob.declare_resource_group()method. We then passg.ofileas the output file root to run_gwas.py as that represents the temporary file root given to all files in the resource group ({root} when declaring the resource group).
Clumping By Chromosome
The second function performs clumping for a given chromosome. The input arguments are the Batch
for which to create a new BashJob, the PLINK binary file root, the association results
with at least two columns (SNP and P), and the chromosome for which to do the clumping for.
The return value is the new BashJob created.
def clump(batch, bfile, assoc, chr):
"""
Clump association results with PLINK
"""
c = batch.new_job(name=f'clump-{chr}')
c.image('hailgenetics/genetics:0.2.37')
c.memory('1Gi')
c.command(f'''
plink --bfile {bfile} \
--clump {assoc} \
--chr {chr} \
--clump-p1 0.01 \
--clump-p2 0.01 \
--clump-r2 0.5 \
--clump-kb 1000 \
--memory 1024
mv plink.clumped {c.clumped}
''')
return c
A couple of things to note about this function:
We use the image
hailgenetics/geneticswhich is a publicly available Docker image from Docker Hub maintained by the Hail team that contains many useful bioinformatics tools including PLINK.We explicitly tell PLINK to only use 1Gi of memory because PLINK defaults to using half of the machine’s memory. PLINK’s memory-available detection mechanism is unfortunately unaware of the memory limit imposed by Batch. Not specifying resource requirements correctly can cause performance degradations with PLINK.
PLINK creates a hard-coded file plink.clumped. We have to move that file to a temporary Batch file {c.clumped} in order to use that file in downstream jobs.
Merge Clumping Results
The third function concatenates all of the clumping results per chromosome into a single file
with one header line. The inputs are the Batch for which to create a new BashJob
and a list containing all of the individual clumping results files. We use the ubuntu:24.04
Docker image for this job. The return value is the new BashJob created.
def merge(batch, results):
"""
Merge clumped results files together
"""
merger = batch.new_job(name='merge-results')
merger.image('ubuntu:24.04')
if results:
merger.command(f'''
head -n 1 {results[0]} > {merger.ofile}
for result in {" ".join(results)}
do
tail -n +2 "$result" >> {merger.ofile}
done
sed -i -e '/^$/d' {merger.ofile}
''')
return merger
Control Code
The last thing we want to do is use the functions we wrote above to create new jobs
on a Batch which can be executed with the ServiceBackend.
First, we import the Batch module as hb.
import hailtop.batch as hb
Next, we create a Batch specifying the backend is the ServiceBackend
and give it the name ‘clumping’.
backend = hb.ServiceBackend()
batch = hb.Batch(backend=backend, name='clumping')
We create InputResourceFile objects for the VCF file and
phenotypes file using the Batch.read_input() method. These
are the inputs to the entire Batch and are not outputs of a BashJob.
vcf = batch.read_input('gs://hail-tutorial/1kg.vcf.bgz')
phenotypes = batch.read_input('gs://hail-tutorial/1kg_annotations.txt')
We use the gwas function defined above to create a new job on the batch to
perform a GWAS that outputs a binary PLINK file and association results:
g = gwas(batch, vcf, phenotypes)
We call the clump function once per chromosome and aggregate a list of the
clumping results files passing the outputs from the g job defined above
as inputs to the clump function:
results = []
for chr in range(1, 23):
c = clump(batch, g.ofile, g.ofile.assoc, chr)
results.append(c.clumped)
Finally, we use the merge function to concatenate the results into a single file
and then write this output to a permanent location using Batch.write_output().
The inputs to the merge function are the clumped output files from each of the clump
jobs.
m = merge(batch, results)
batch.write_output(m.ofile, 'gs://<MY_BUCKET>/batch-clumping/1kg-caffeine-consumption.clumped')
The last thing we do is submit the Batch to the service and then close the Backend:
batch.run(open=True, wait=False) # doctest: +SKIP
backend.close()
Synopsis
We provide the code used above in one place for your reference:
import argparse
import hail as hl
def run_gwas(vcf_file, phenotypes_file, output_file):
table = hl.import_table(phenotypes_file, impute=True).key_by('Sample')
hl.import_vcf(vcf_file).write('tmp.mt')
mt = hl.read_matrix_table('tmp.mt')
mt = mt.annotate_cols(pheno=table[mt.s])
mt = hl.sample_qc(mt)
mt = mt.filter_cols((mt.sample_qc.dp_stats.mean >= 4) & (mt.sample_qc.call_rate >= 0.97))
ab = mt.AD[1] / hl.sum(mt.AD)
filter_condition_ab = (
(mt.GT.is_hom_ref() & (ab <= 0.1))
| (mt.GT.is_het() & (ab >= 0.25) & (ab <= 0.75))
| (mt.GT.is_hom_var() & (ab >= 0.9))
)
mt = mt.filter_entries(filter_condition_ab)
mt = hl.variant_qc(mt)
mt = mt.filter_rows(mt.variant_qc.AF[1] > 0.01)
eigenvalues, pcs, _ = hl.hwe_normalized_pca(mt.GT)
mt = mt.annotate_cols(scores=pcs[mt.s].scores)
gwas = hl.linear_regression_rows(
y=mt.pheno.CaffeineConsumption,
x=mt.GT.n_alt_alleles(),
covariates=[1.0, mt.pheno.isFemale, mt.scores[0], mt.scores[1], mt.scores[2]],
)
gwas = gwas.select(SNP=hl.variant_str(gwas.locus, gwas.alleles), P=gwas.p_value)
gwas = gwas.key_by(gwas.SNP)
gwas = gwas.select(gwas.P)
gwas.export(f'{output_file}.assoc', header=True)
hl.export_plink(mt, output_file, fam_id=mt.s, ind_id=mt.s)
if __name__ == '__main__':
parser = argparse.ArgumentParser()
parser.add_argument('--vcf', required=True)
parser.add_argument('--phenotypes', required=True)
parser.add_argument('--output-file', required=True)
parser.add_argument('--cores', required=False)
args = parser.parse_args()
if args.cores:
hl.init(master=f'local[{args.cores}]')
run_gwas(args.vcf, args.phenotypes, args.output_file)
FROM hailgenetics/hail:0.2.37
COPY run_gwas.py /
import hailtop.batch as hb
def gwas(batch, vcf, phenotypes):
"""
QC data and get association test statistics
"""
cores = 2
g = batch.new_job(name='run-gwas')
g.image('us-docker.pkg.dev/<MY_PROJECT>/1kg-gwas:latest')
g.cpu(cores)
g.declare_resource_group(
ofile={'bed': '{root}.bed', 'bim': '{root}.bim', 'fam': '{root}.fam', 'assoc': '{root}.assoc'}
)
g.command(f"""
python3 /run_gwas.py \
--vcf {vcf} \
--phenotypes {phenotypes} \
--output-file {g.ofile} \
--cores {cores}
""")
return g
def clump(batch, bfile, assoc, chr):
"""
Clump association results with PLINK
"""
c = batch.new_job(name=f'clump-{chr}')
c.image('hailgenetics/genetics:0.2.37')
c.memory('1Gi')
c.command(f"""
plink --bfile {bfile} \
--clump {assoc} \
--chr {chr} \
--clump-p1 0.01 \
--clump-p2 0.01 \
--clump-r2 0.5 \
--clump-kb 1000 \
--memory 1024
mv plink.clumped {c.clumped}
""")
return c
def merge(batch, results):
"""
Merge clumped results files together
"""
merger = batch.new_job(name='merge-results')
merger.image('ubuntu:24.04')
if results:
merger.command(f"""
head -n 1 {results[0]} > {merger.ofile}
for result in {" ".join(results)}
do
tail -n +2 "$result" >> {merger.ofile}
done
sed -i -e '/^$/d' {merger.ofile}
""")
return merger
if __name__ == '__main__':
backend = hb.ServiceBackend()
batch = hb.Batch(backend=backend, name='clumping')
vcf = batch.read_input('gs://hail-tutorial/1kg.vcf.bgz')
phenotypes = batch.read_input('gs://hail-tutorial/1kg_annotations.txt')
g = gwas(batch, vcf, phenotypes)
results = []
for chr in range(1, 23):
c = clump(batch, g.ofile, g.ofile.assoc, chr)
results.append(c.clumped)
m = merge(batch, results)
batch.write_output(m.ofile, 'gs://<MY_BUCKET>/batch-clumping/1kg-caffeine-consumption.clumped')
batch.run(open=True, wait=False)
backend.close()