Hail Query
Simplified Analysis
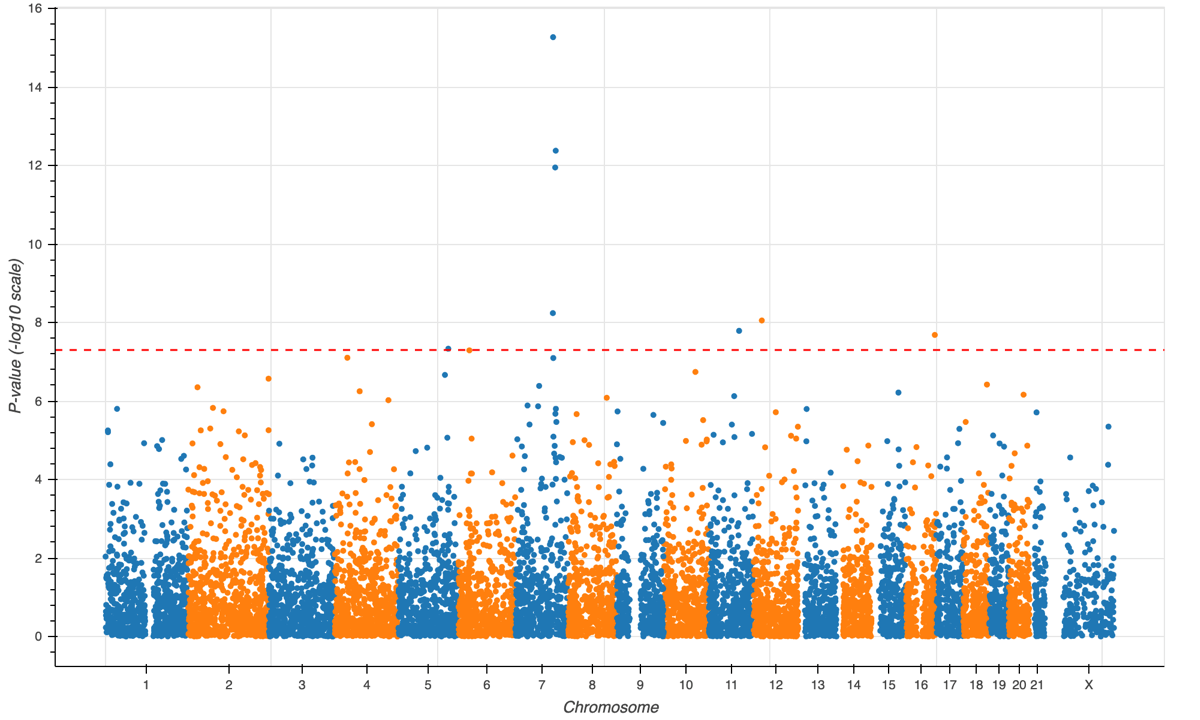
Hail Query provides powerful, easy-to-use data science tools. Interrogate data at every scale: small datasets on a laptop through to biobank-scale datasets (e.g. UK Biobank, gnomAD, TopMed, FinnGen, and Biobank Japan) in the cloud.
Genomic Dataframes
Modern data science is driven by numeric matrices (see Numpy) and tables (see R dataframes and Pandas). While sufficient for many tasks, none of these tools adequately capture the structure of genetic data. Genetic data combines the multiple axes of a matrix (e.g. variants and samples) with the structured data of tables (e.g. genotypes). To support genomic analysis, Hail introduces a powerful and distributed data structure combining features of matrices and dataframes called MatrixTable.
Input Unification
The Hail MatrixTable unifies a wide range of input formats (e.g. vcf, bgen, plink, tsv, gtf, bed files), and supports scalable queries, even on petabyte-size datasets. Hail's MatrixTable abstraction provides an integrated and scalable analysis platform for science.